
Khoi Bui Dinh
![]()
![]()
![]()
ITS, 16S, 18S: Special regions in bacterial and fungal genome
Jan 23rd, 2026
Warning: a messy and could-be-more-structured attempt at explaining bacterial genomes for myself.
The first time I was doing a bioinformatics service using Oxford Nanopore Sequencing (ONT) full length was sometimes late in 2025. I had never worked with ONT full length before, nor had I did any work with fungal species classification. So naturally, now that the service is delivered, I had the chance to sit back and learn a bit more about these special regions, and what made them viable for taxonomic classification.
DNA barcoding, what is it?
-
So genomes are just collection of genes yes? And each gene is consists of many nucleotide. DNA barcoding refer to the usage of a small fragment of DNA to characterise the species, with the idea that each unique species must contains a region (or regions) able for classification. So homo sapiens (human) and pan troglodytes (chimpanzees) share something of 98% of identical genomes, but those 2% makes up the differences. So by using something in those 2%, we can tell which is which!
-
DNA barcoding is more likely to used in microbial studies though, where we can’t tell the differences just by looking, and bacteria for example, share an even larger similarity. Hence, DNA barcodes need to be small regions of a few hundred bp (from 400 - 800 bp 1), and they need to be very highly conserved. As these regions are highly conserved, they are almost identical in organisms of the same species.
-
Take the following example: E.Coli being one of the very well studied (I suppose) bacteria, and it contains a 16S region (more on this later), where if mapped to many reference genomes may allow us to know that it is in fact, E.Coli. If this region however is mapped almost entirely to E.Coli, but differs a bit, it may call for further experiments before deciding whether these are E.Coli strains. From what I’ve gathered, there are strict benchmarks to decide whether it’s a new strain of bacteria or not.
Okay but then, which region(s) should we use?
-
Good question, ha! So I was processing 16S / ITS rRNA sequencing data of some water samples. The technology uses, as the name suggests, 16S rRNA regions to identify bacteria and ITS regions to identify fungi. But why 16S for bacteria? What is ITS? Do fungi have 16S region? Why not the same for both? So to the rabbit hole I went…
- I came across Kress & Erickson, 20081, apparently this idea was already around when I was 8. The authors pointed out 3 criteria for selecting a gene region as DNA barcode:
(i) contain significant species-level genetic variability and divergence
(ii) possess conserved flanking sites for developing universal PCR primers for wide taxonomic application, and
(iii) have a short sequence length so as to facilitate current capabilities of DNA extraction and amplification. -
The first criteria seems intuitive, if the species’s “identifying gene region” is just the same with other species, then we need another gene region ._. So is the same with the second criteria, imagine trying to identify a gene region A ranging 43 bp from location 1,321,234 to location 1,321,277 of a chromosome 12 only to discover that this A region starts from 3,123,421 instead! (This is a make up example of course) Then we won’t be able to develop a consistent primer for PCR these gene regions. And finally, I’d like to think of this as “the longer the region we have to sequence, the higher the cost of our science”. Of course this paper was in 2008, the data that I worked with in 2025 is already full-length of 16S regions, so they are way longer than what was available then.
- So by then our knowledge of bacterial rRNA regions were already quite established: in ribosomes there are two subunits, SSU (small one) and LSU (large one), and these subunits contain highly conserved flanking sites, such as 16S, 18S.
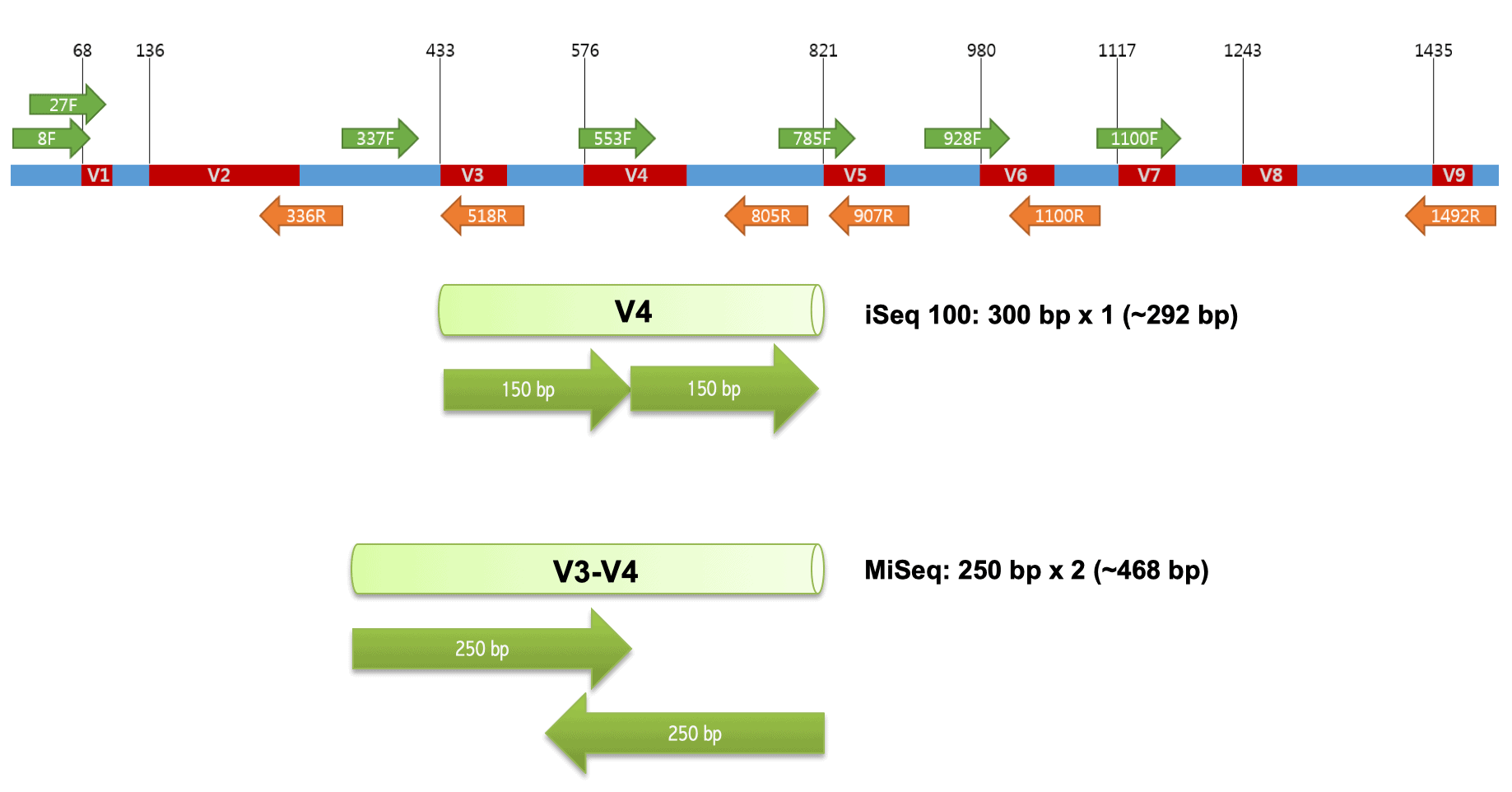
-
16S happens to contain a highly conserved primer binding hotspots, so your universal PCR primers would work well in extracting these regions. They also contains something called the hypervariable regions (V1-V9), these regions contain very diverse sequences, allow for differentiation between species in a mixed sample. That’s why it’s being used a lot!
-
Chakravorty et al, 2007 proposed using 16S gene segments to diagnose of pathogenic bacteria. He demonstrated the effectiveness of using regions V2, V3 and V6 for taxonomic classification. I remembered a friend of mine doing her capstone project in 2024 used V3-V4 regions for her sequencing. I have yet to look deeper into which one is better (?!), but Johnson, J.S., Spakowicz, D.J., Hong, BY. et al, 2019 3 seemed to have their answers. They noted that using full length 16S sequencing, meaning that you sequence the entire V1-V9, has more “real and significant” advantages over targeting specific regions, despite their possible errors.
I really need to go to study...
- As a Computer Science student (well alumni now), there’s an obvious gap in biology knowledge that needs to be filled in. In the meantime, I’m juggling between work and self-learn in some down time, but I do hope to have an opportunity to take some biology courses, think way deeper about the science that I’m doing (why this tool and not that?), or know more about the beginning of my projects rather than just “take the FASTQs and process”.
- Well, I’m still happy though. Happy that once in a while I might get to visit these posts and add more grounded knowledge :>
Citations
-
Kress, W. J., & Erickson, D. L. (2008). DNA barcodes: genes, genomics, and bioinformatics. Proceedings of the National Academy of Sciences of the United States of America, 105(8), 2761–2762. https://doi.org/10.1073/pnas.0800476105
-
Magray, M. S., Kumar, A., Rawat, A. K., & Srivastava, S. (2011). Identification of Escherichia coli through analysis of 16S rRNA and 16S-23S rRNA internal transcribed spacer region sequences. Bioinformation, 6(10), 370–371. https://doi.org/10.6026/97320630006370
-
Johnson, J.S., Spakowicz, D.J., Hong, BY. et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun 10, 5029 (2019). https://doi.org/10.1038/s41467-019-13036-1